MLST-Multilocus Sequence Typing
Overview
After taxonomic classification, we can identify the sequence type of a bacterial genome using MLST.
MLST stands for multilocus sequence typing. It compares specific housekeeping genes against known allele databases and assigns a sequence type when a matching profile is found.
In this lesson, we will run mlst on the filtered assembly file and save the result inside the classify directory.
Learning objectives
By the end of this lesson, you should be able to:
- explain what MLST is
- activate the
mlstenvironment - run
mlston an assembled genome - save MLST results into the
classifydirectory - interpret the basic MLST output
Input file
In the previous lessons, we created a filtered assembly file:
assemblies/S1_contigs_filtered.fastaCheck that the file exists:
ls assembliesYou should see:
S1_contigs_filtered.fastaFor this lesson, we will use the filtered assembly file:
assemblies/S1_contigs_filtered.fastaCreate output directory
We will save the MLST result inside the classify directory.
Check that the directory exists from Kraken2 analysis:
lsActivate the MLST environment
Activate the mlst environment:
conda activate mlstCheck that mlst is available:
mlst --versionyou will see:
mlst 2.33.1Run MLST
Run mlst on the filtered assembly:
mlst assemblies/S1_contigs_filtered.fasta > classify/S1_mlst.txtUnderstanding the MLST command
| Part | Meaning |
|---|---|
mlst |
Runs the MLST tool |
assemblies/S1_contigs_filtered.fasta |
Input filtered assembly file |
> |
Redirects the output to a file |
classify/S1_mlst.txt |
Output MLST result file |
Check MLST output
List the classify directory:
ls classifyYou should see:
S1_mlst.txtView the MLST result:
cat classify/S1_mlst.txtExample MLST result
After running:
cat classify/S1_mlst.txtyou may see output like this:
assemblies/S1_contigs_filtered.fasta vcholerae 69 adk(7) gyrB(11) mdh(4) metE(37) pntA(12) purM(1) pyrC(20)Interpreting this result
| Field | Value | Meaning |
|---|---|---|
| Input file | assemblies/S1_contigs_filtered.fasta |
Assembly file used for MLST |
| Scheme | vcholerae |
Vibrio cholerae MLST scheme |
| Sequence type | 69 |
Assigned MLST sequence type |
adk |
7 |
Allele 7 for the adk gene |
gyrB |
11 |
Allele 11 for the gyrB gene |
mdh |
4 |
Allele 4 for the mdh gene |
metE |
37 |
Allele 37 for the metE gene |
pntA |
12 |
Allele 12 for the pntA gene |
purM |
1 |
Allele 1 for the purM gene |
pyrC |
20 |
Allele 20 for the pyrC gene |
This result means that sample S1 was assigned to the Vibrio cholerae MLST scheme and identified as ST69:
What is the MLST sequence type of sample S1?
The MLST sequence type is:
ST69The MLST scheme is:
vcholeraeSave and organize results
Your classification folder should now contain results from taxonomy and MLST.
Check the folder:
ls classifyYou may see files such as:
S1.kraken2.output
S1.kraken2.report
S1.krona
S1.krona.html
S1_mlst.txtDeactivate the classify environment:
conda deactivateDownload MLST result from the server
If you are working on a remote server, download the MLST result to your local computer.
From your local desktop terminal, run:
scp genomevm@172.28.28.12:~/tanzim/classify/S1_mlst.txt .This downloads S1_mlst.txt to your current local folder.
Open S1_mlst.txt manually using Notepad, Notepad++, Excel, or VS Code.
Use:
open S1_mlst.txtUse:
xdg-open S1_mlst.txtCompare taxonomy and MLST
Taxonomic classification and MLST provide different but complementary information.
| Analysis | Tool | Main output |
|---|---|---|
| Taxonomic classification | Kraken2 | Likely organism identity |
| Interactive taxonomy plot | Krona | Visual taxonomic hierarchy |
| Sequence typing | MLST | Sequence type based on allele profile |
Do your Kraken2 and MLST results agree?
For example, does the MLST scheme match the organism suggested by Kraken2?
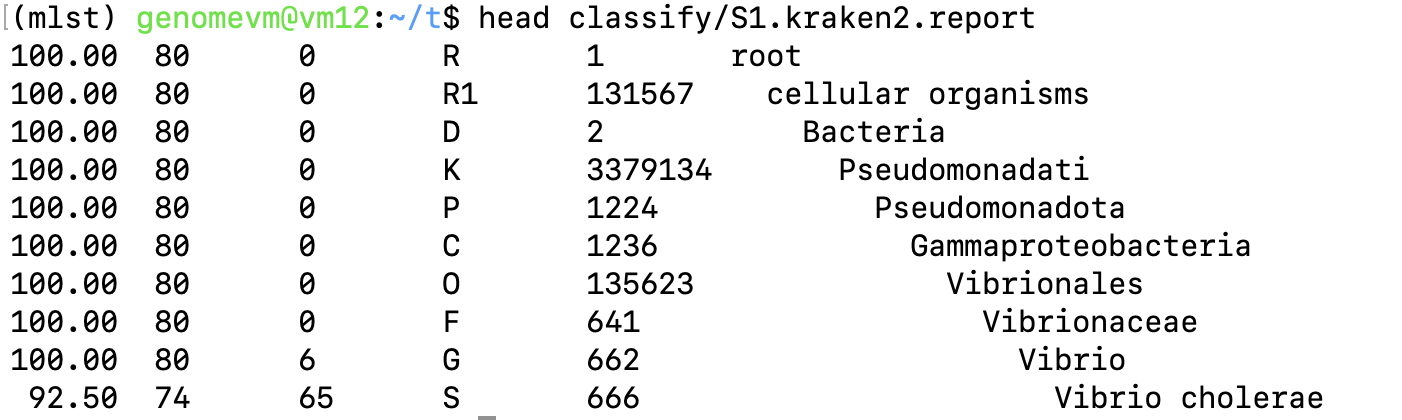
 Taxonomy: Kraken2 vs MLST scheme
Taxonomy: Kraken2 vs MLST scheme
The results should generally agree. For example, if Kraken2 suggests the sample is Vibrio cholerae, the MLST tool should ideally use an vcholerae MLST scheme. If the scheme and taxonomy do not match, the sample identity or classification should be checked carefully.
Importance of Vibrio cholerae ST69

Final directory structure
At the end of this lesson, your directory should look like this:
.
├── assemblies
│ ├── S1_contigs.fasta
│ └── S1_contigs_filtered.fasta
└── classify
├── S1.kraken2.output
├── S1.kraken2.report
├── S1.krona
├── S1.krona.html
└── S1_mlst.txtPractical exercise
Complete the following tasks:
- Activate the
mlstenvironment. - Run
mlstonS1_contigs_filtered.fasta. - Save the result as
classify/S1_mlst.txt. - Open the MLST result.
- Record the MLST scheme and sequence type.
- Compare the MLST result with the Kraken2 taxonomy result.
Create a small table in your notes:
| Sample | Kraken2 main assignment | MLST scheme | Sequence type |
|---|---|---|---|
| S1 |
Fill in the table using:
classify/S1.kraken2.report
classify/S1_mlst.txtKey points
- MLST assigns a sequence type using housekeeping gene allele profiles.
- The input is the filtered assembly file.
- The output is saved as
classify/S1_mlst.txt. - MLST results can be compared with Kraken2 taxonomy results.
- If taxonomy and MLST disagree, the sample should be checked carefully.
Next step
In the next lesson, we will annotate the filtered assembly file using Prokka.