flowchart LR F[drawing room] --> G[bedroom 1/ environment 1/ python 2.7] F --> H[bedroom 2/ environment 2/ python 3.7]
Workflow Management Systems
Why autmation is needed in bioinformatics analysis?
Working as a bioinformatician is cumbersome and stressful unless you become a bioinformagician, and how you can be a bioinformagician? Simple, all you have to do is automate your tasks, write scripts, make it stable, scalable and reproducible. Once done, give a command to your computer, wait and chill until it finishes.

What is automation?
Suppose you are an Finance executive in a corporate office. Your office has 1000 employees who comes at 9 am, punch his finger as in and punch again at 6 pm as out. You manager asked you to make salary sheet for all employees and he need it within 2 days! You can’t say a word because a boss is boss! Now, you started to take notes of
- number of working days in that month
- number of paid leaves
- number of unpaid leaves
- number of working hours per day
- number of hours as overtime
- whether a fixed term employee or contractual or daily wager
- festive bonus based on months s/he is working
Imagine your life if you have to do everything manually! and that’s not just for one employee, you have 999 others!!!
Now what can do is:
- write a programming script that will automatically calculate everything based on punch in and out for each day and at the end calculate salary.
What more you can do is write another script that will calculate simultaneously (side by side) for each employee and provide a summary for all 1000 employees.
That’s it! Your taks is finished with in a day. What you did is just wirte a bunch codes and automated the whole process.
Automation in bioinformatics
As a bioinformatician you need to learn at least either Python, R or Perl and Bash programming language. You can automate a process, say for example, bacterial genomics starting from data quality check, trimming, assembly, assembly assessment, taxonomic classification, annotation of genome etc. etc. You can write a script either on bashor R or python which will do all these processes one by one of all samples. That’s it! What can go wrong! right?
While you are writing and testing the script, you observed that a software is working fine but another one is not. You looked for the errors and found that software A is written in Python version 2.7 but software B is written in Python version 3.7. That means A is dependent of python 2.7 and B is dependent on python 3.7. Since these two are different in syntax, one software is giving error while other one is working fine.
The solution is you make partition or an environment where A will be installed with Python 2.7 and another environment where B will be installed with Python 3.7. You can think of it as 2 bed and a drawing room flat, where each bedroom is environment has separated door to enter.
This environemtents are created either with Docker or singularity or conda.
Conda, Singularity, Docker?
There are three solutions for managing software dependencies:
- Docker is a software that allows to package a small virtual operating system (or a “container”) containing all the software and data necessary for running an analysis.
- Singularity also creates software containers, similarly to Docker. However, it can more easily interface with the user’s filesystem, without the need to have special permissions.
- Conda is a package manager, also very popular in bioinformatics. Instead of creating virtual OS containers, Conda instead creates software environments (think of them as directories) where all the software is locally installed, including all its dependencies. The use of individual environments ensures that software packages with incompatible dependencies can still be used within the same pipeline.
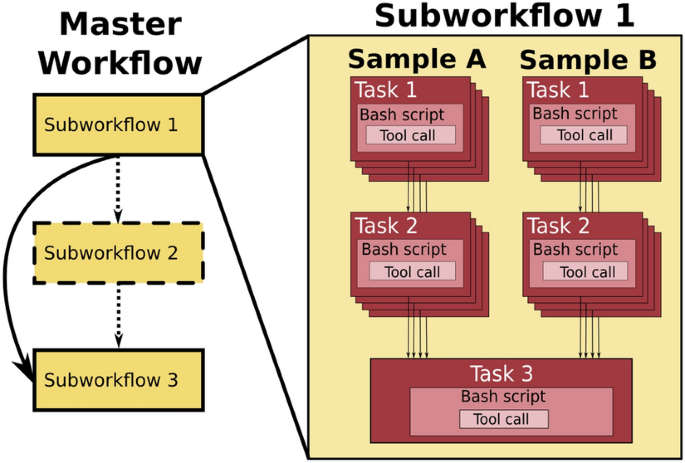
Workflows
Analysing data involves a sequence of tasks, including gathering, cleaning, and processing data. These sequence of tasks are called a workflow or a pipeline. These workflows typically require executing multiple software packages, sometimes running on different computing environments, such as a desktop or a compute cluster. Traditionally these workflows have been joined together in scripts using general purpose programming languages such as Bash or Python.
However, as workflows become larger and more complex, the management of the programming logic and software becomes difficult.
Workflow management systems
Workflow Management Systems (WfMS), such as Snakemake, Galaxy, and Nextflow have been developed specifically to manage computational data-analysis workflows in fields such as Bioinformatics, Imaging, Physics, and Chemistry.
WfMS contain multiple features that simplify the development, monitoring, execution and sharing of pipelines.
Key features include;
- Run time management: Management of program execution on the operating system and splitting tasks and data to run at the same time in a process called parallelisation.
- Software management: Use of technology like containers, such as Docker or Singularity, that packages up code and all its dependencies so the application runs reliably from one computing environment to another.
- Portability & Interoperability: Workflows written on one system can be run on another computing infrastructure e.g., local computer, compute cluster, or cloud infrastructure.
- Reproducibility: The use of software management systems and a pipeline specification means that the workflow will produce the same results when re-run, including on different computing platforms.
- Reentrancy: Continuous checkpoints allow workflows to resume from the last successfully executed steps.