Trimming to Retain High Quality Data
Overview
After checking raw read quality with FastQC, we may need to trim reads to remove low-quality bases or unwanted sequence at the beginning or end of reads.
In this lesson, we will use Trimmomatic to trim paired-end reads and then run FastQC again to check whether read quality has improved.
Learning objectives
By the end of this lesson, you should be able to:
- explain why read trimming may be required
- run
Trimmomaticon paired-end FASTQ files - identify paired and unpaired trimming outputs
- run
FastQCon trimmed reads - organize raw reads, trimmed reads, and QC reports into folders
Activate the QC environment
If you are continuing from the previous lesson, you may already be inside the qc environment.
If not, activate it first:
conda activate qcThe terminal will appear as
(qc) genomevm@genome-clone-vm2:~tanzim$Check that trimmomatic is available:
trimmomatic -versionThe terminal will appear as
(qc) genomevm@genome-clone-vm2:~tanzim$
0.40Check that fastqc is also available:
fastqc --versionThe terminal will appear as
(qc) genomevm@genome-clone-vm2:~tanzim$
Fastqc v0.12.1Input files
For this lesson, we will use paired-end FASTQ files from sample S1.
S1_R1.fastq.gz
S1_R2.fastq.gzCheck that the files are present:
lsCreate output folders
Before running trimming, create separate folders for trimmed reads and QC reports.
mkdir -p trimmed_reads
mkdir -p qc_reportsRun Trimmomatic
Run Trimmomatic in paired-end mode:
trimmomatic PE \
S1_R1.fastq.gz S1_R2.fastq.gz \
trimmed_reads/S1_trim_R1.fastq.gz trimmed_reads/S1_unpaired_R1.fastq.gz \
trimmed_reads/S1_trim_R2.fastq.gz trimmed_reads/S1_unpaired_R2.fastq.gz \
SLIDINGWINDOW:4:20 LEADING:3 TRAILING:3 HEADCROP:15 MINLEN:40Understanding the command
| Part | Meaning |
|---|---|
PE |
Paired-end mode |
S1_R1.fastq.gz |
Input forward reads |
S1_R2.fastq.gz |
Input reverse reads |
S1_trim_R1.fastq.gz |
Trimmed paired forward reads |
S1_unpaired_R1.fastq.gz |
Forward reads whose pair was removed |
S1_trim_R2.fastq.gz |
Trimmed paired reverse reads |
S1_unpaired_R2.fastq.gz |
Reverse reads whose pair was removed |
SLIDINGWINDOW:4:20 |
Scan with a 4-base window and cut when average quality falls below 20 |
LEADING:3 |
Remove low-quality bases from the beginning |
TRAILING:3 |
Remove low-quality bases from the end |
HEADCROP:15 |
Remove the first 15 bases from each read |
MINLEN:40 |
Remove reads shorter than 40 bases after trimming |
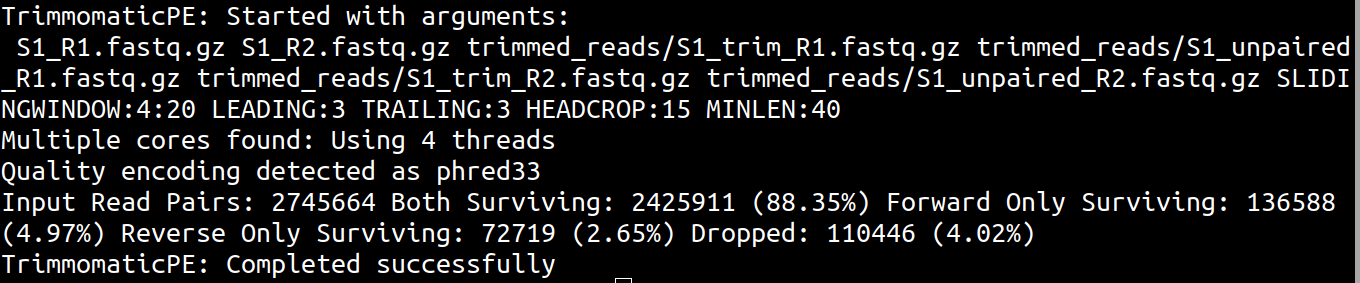
For downstream paired-end genome assembly, we usually use the paired trimmed files:
trimmed_reads/S1_trim_R1.fastq.gz
trimmed_reads/S1_trim_R2.fastq.gzThe unpaired files can be kept or removed depending on the analysis plan.
Check trimming outputs
List the trimmed read files:
ls trimmed_readsYou should see:
S1_trim_R1.fastq.gz
S1_trim_R2.fastq.gz
S1_unpaired_R1.fastq.gz
S1_unpaired_R2.fastq.gzRun FastQC on trimmed reads
Now run FastQC again on the trimmed paired reads:
fastqc -t 4 \
trimmed_reads/S1_trim_R1.fastq.gz \
trimmed_reads/S1_trim_R2.fastq.gz \
-o qc_reportsCheck the output:
ls qc_reportsYou should see new FastQC reports for the trimmed reads:
S1_trim_R1_fastqc.html
S1_trim_R1_fastqc.zip
S1_trim_R2_fastqc.html
S1_trim_R2_fastqc.zipOpen the .html files manually from File Explorer.
Use:
open S1_trim_R1_fastqc.html
open S1_trim_R2_fastqc.htmlUse:
xdg-open S1_trim_R1_fastqc.html
xdg-open S1_trim_R2_fastqc.htmlCompare raw and trimmed reads
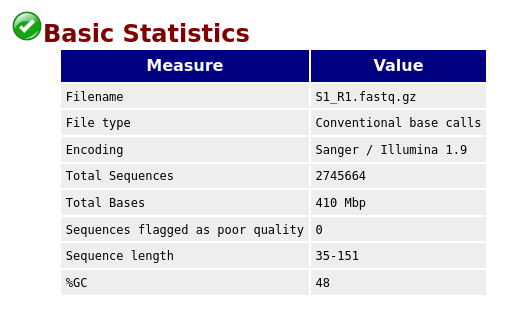
Compare the original FastQC reports with the trimmed FastQC reports. Focus on:
- summary statistics
- per base sequence quality
- per sequence quality scores
Compare S1_R1_fastqc.html with S1_trim_R1_fastqc.html. What changed after trimming?
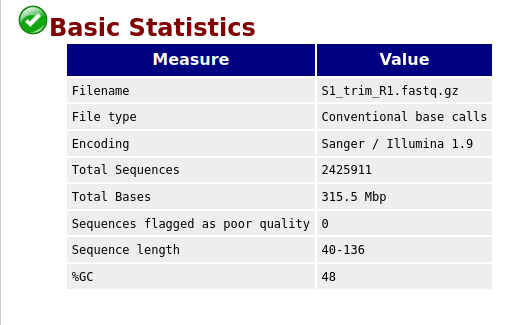
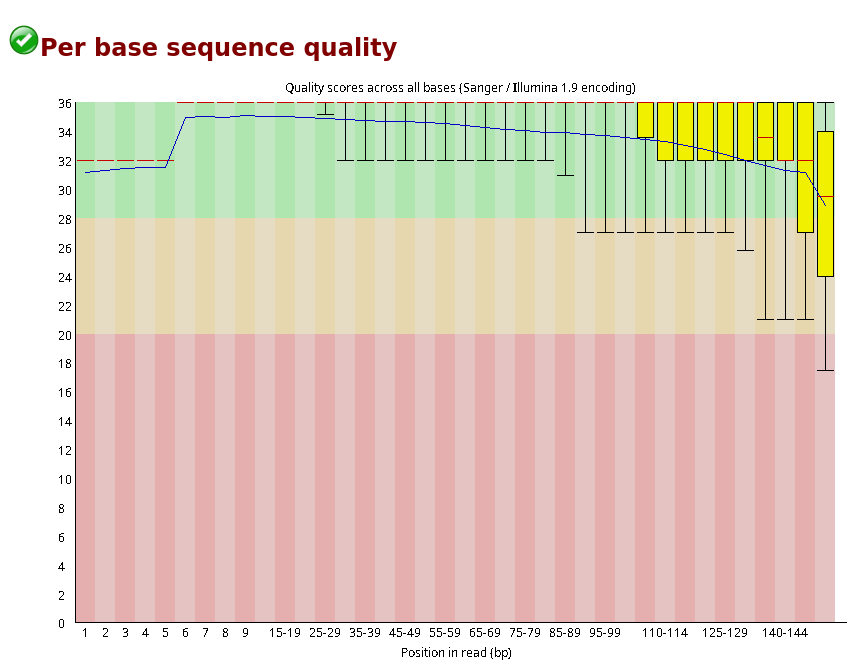
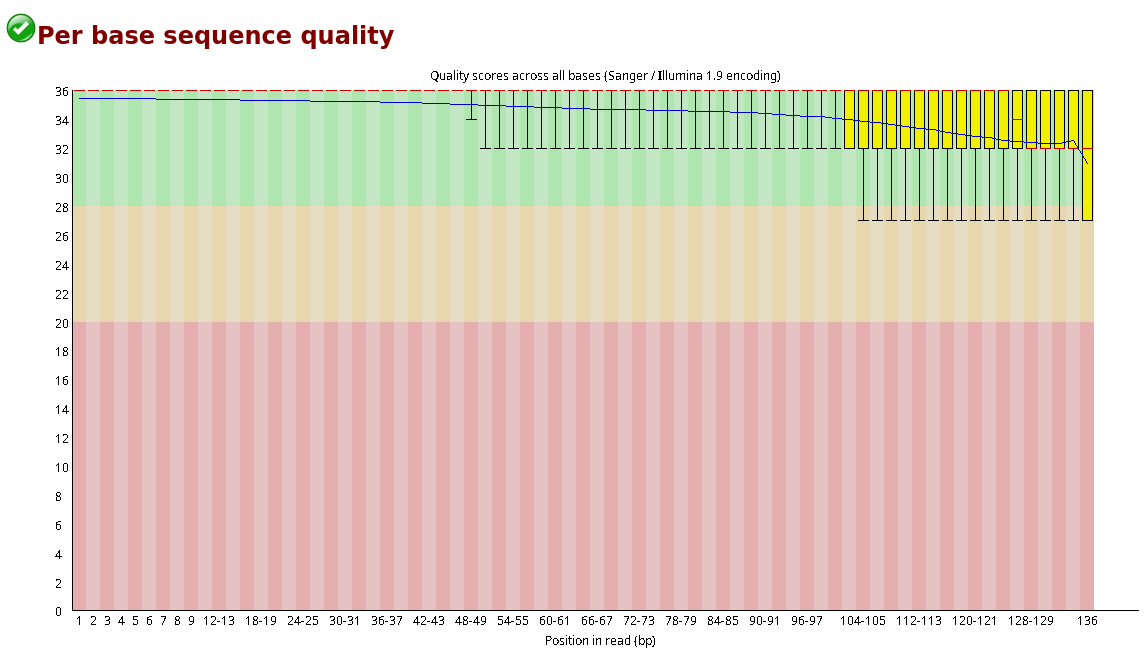
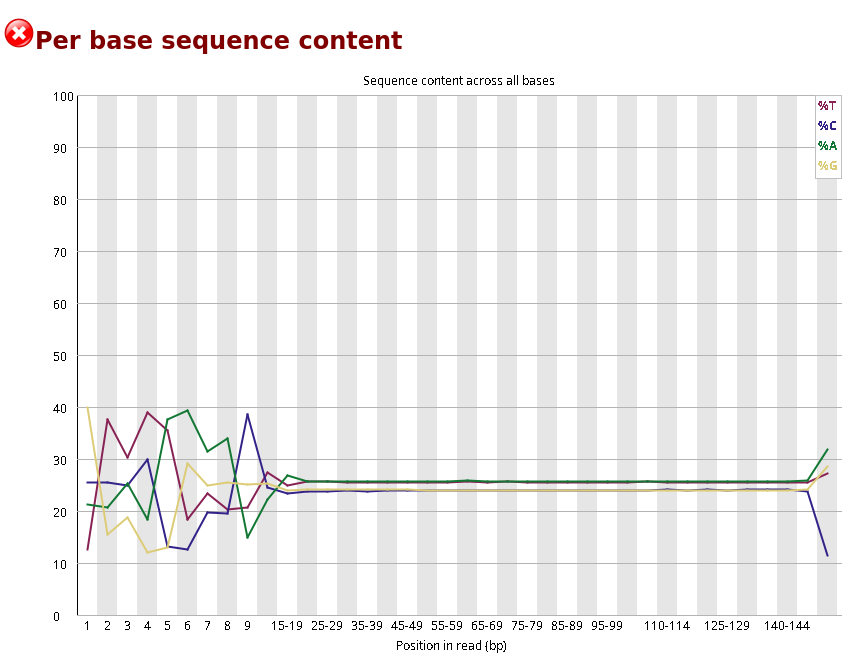
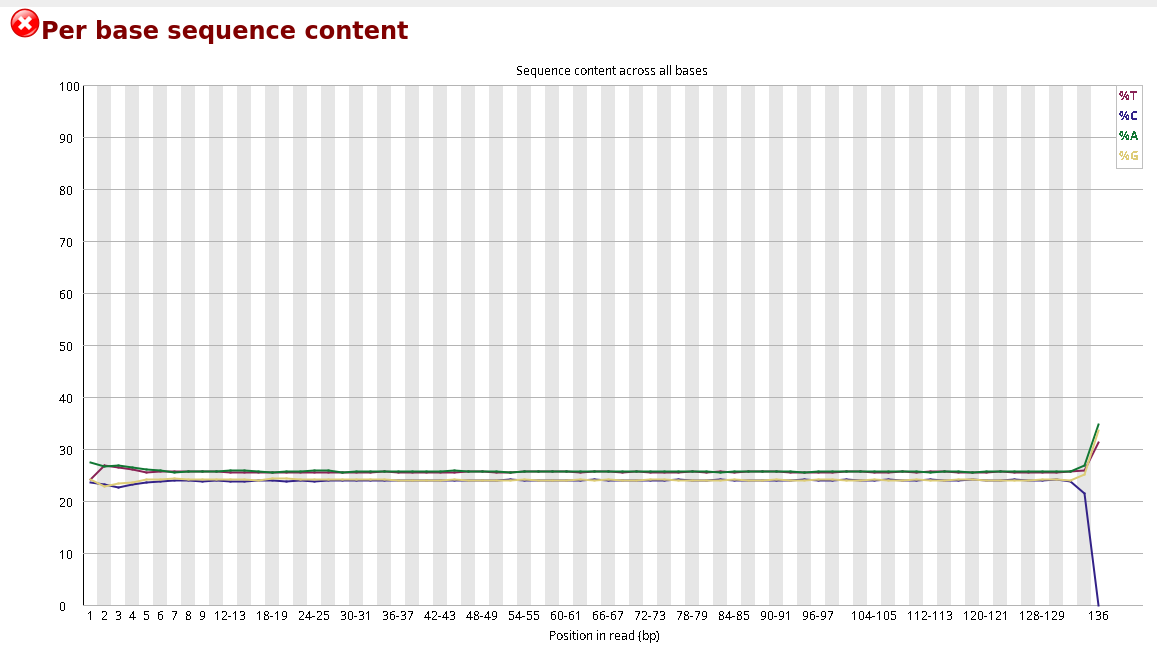 Per base sequence quality before and after trimming bases
Per base sequence quality before and after trimming bases
Organize files
Now we will organize the working directory. Create three folders:
mkdir -p raw_reads trimmed_reads qc_reportsMove the original raw reads into raw_reads:
mv S1_R1.fastq.gz S1_R2.fastq.gz raw_reads/If any FastQC reports were created in the current directory, move them to qc_reports:
mv *fastqc* qc_reports/The trimmed reads should already be inside trimmed_reads.
Check your folder structure:
lsExpected folders:
raw_reads
trimmed_reads
qc_reportsCheck inside each folder:
ls raw_reads
ls trimmed_reads
ls qc_reportsIf your instructor tells you that unpaired reads are not needed, remove them:
rm trimmed_reads/*unpaired*Final directory structure At the end of this lesson, your directory should look like this:
.
├── raw_reads
│ ├── S1_R1.fastq.gz
│ └── S1_R2.fastq.gz
├── trimmed_reads
│ ├── S1_trim_R1.fastq.gz
│ └── S1_trim_R2.fastq.gz
└── qc_reports
├── S1_trim_R1_fastqc.html
├── S1_trim_R1_fastqc.zip
├── S1_trim_R2_fastqc.html
└── S1_trim_R2_fastqc.zipTrimming removes low-quality bases and unwanted sequence. Trimmomatic PE is used for paired-end read trimming. Paired trimmed files are usually used for genome assembly. Run FastQC again after trimming. Keep raw reads, trimmed reads, and QC reports in separate folders.