Quality Control of Raw Reads
Overview
Read quality control is the first checkpoint in bacterial whole-genome sequencing analysis. Before genome assembly or downstream analysis, we need to check whether the raw sequencing reads are suitable for analysis.
In this lesson, we will inspect FASTQ files, run quality-control checks, summarise QC reports, and decide whether the reads need trimming.
Learning objectives
By the end of this lesson, you should be able to:
- describe the structure of a FASTQ file
- explain why read quality control is important
- run
FastQCon raw sequencing reads - summarise multiple QC reports using
MultiQC - interpret common QC metrics
- decide whether trimming is needed before assembly
Why do we perform read quality control?
Raw sequencing reads may contain technical problems such as:
- low-quality bases
- adapter contamination
- unusually short reads
- biased base composition
- unexpected GC content
- overrepresented sequences
- possible contamination
If poor-quality reads are used directly, they can affect genome assembly, species identification, antimicrobial resistance gene detection, and phylogenetic analysis.
Quality control helps us decide whether the sequencing data are good enough for downstream analysis.
FASTQ file structure
Short-read sequencing data are usually stored in FASTQ format. A FASTQ record has four lines:
@read_id
ACGTACGTACGTACGT
+
FFFFFFFFFFFFFFFF| Line | Description |
|---|---|
| Line 1 | Read identifier |
| Line 2 | DNA sequence |
| Line 3 | Separator |
| Line 4 | Quality score |
The fourth line contains quality scores. These scores represent confidence in each base call.
Paired-end sequencing files
For paired-end Illumina data, each sample usually has two FASTQ files:
S1_R1.fastq.gz
S1_R2.fastq.gz| File | Meaning |
|---|---|
R1 |
Forward reads |
R2 |
Reverse reads |
.gz |
Compressed file |
For example:
SRR123456_R1.fastq.gz
SRR123456_R2.fastq.gzInspect a FASTQ file
Because FASTQ files are often compressed, use zcat to view them.
zcat S1_R1.fastq.gz | headTo view the first 8 lines:
zcat S1_R1.fastq.gz | head -n 8This should show the first two FASTQ records.
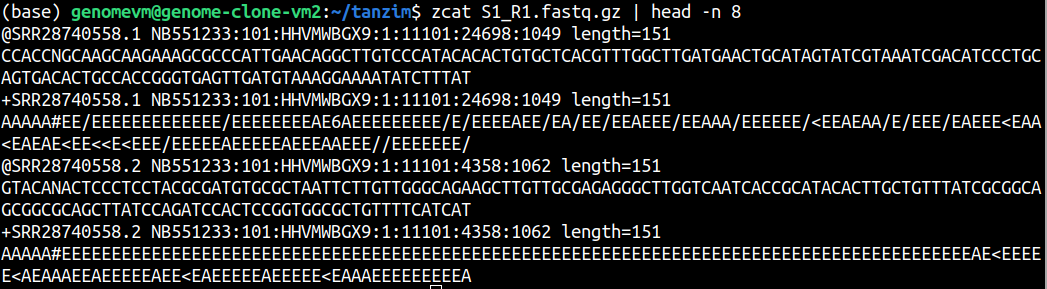
Inspect the first 16 lines of a FASTQ file. How many sequencing reads are shown?
A FASTQ record contains 4 lines. Therefore, 8 lines show 2 sequencing reads.
For example, if the file has 400000 lines, then the number of reads is:
400000 / 4 = 100000 readsYou can calculate this directly:
echo $(( $(zcat S1_R1.fastq.gz | wc -l) / 4 ))2745664Run FastQC
We need to activate the qc environment
conda activate qcNow will run fastqc on sample S1
fastqc -t 4 S1_R1.fastq.gz S1_R2.fastq.gz
Type ls to see the file lists
lsOpen FastQC report
On Windows, open the file manually from the file explorer.
If you are working on your own computer, open the .html file in a browser. On macOS:
open S1_R1_fastqc.htmlOn Linux:
xdg-open S1_R1_fastqc.htmlIn a similar way, open the S1_R2_fastqc.html
Understand the fastqc results
Once you open the html file you will see a summary information with. What important here are
Basic StatisticsPer base sequence qualityPer base sequence content
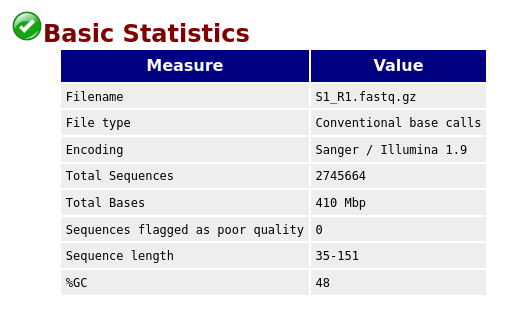
Here,
- number of reads is
2745664 - sequence length
35-151 - %GC
48
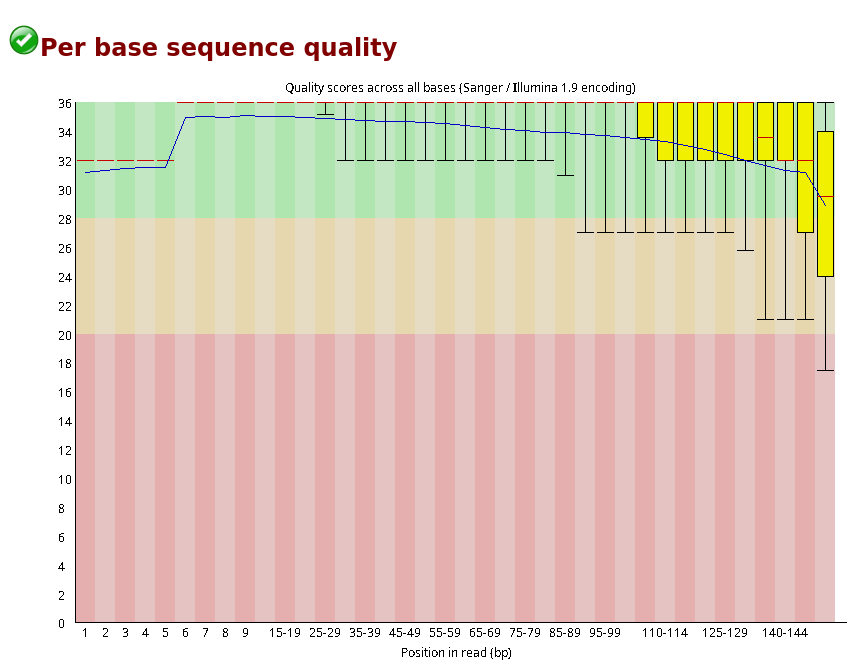 Here,
Here, green is best, yellow is good and red is low quality bases. We can see the average quality score dropping at the end of each read. So we need to trim these bases to keep all the reads above Phred score 20.
Also we need to consider the report of Per base sequence content as some bases are generated randomly, which alternatively we call machine noises.
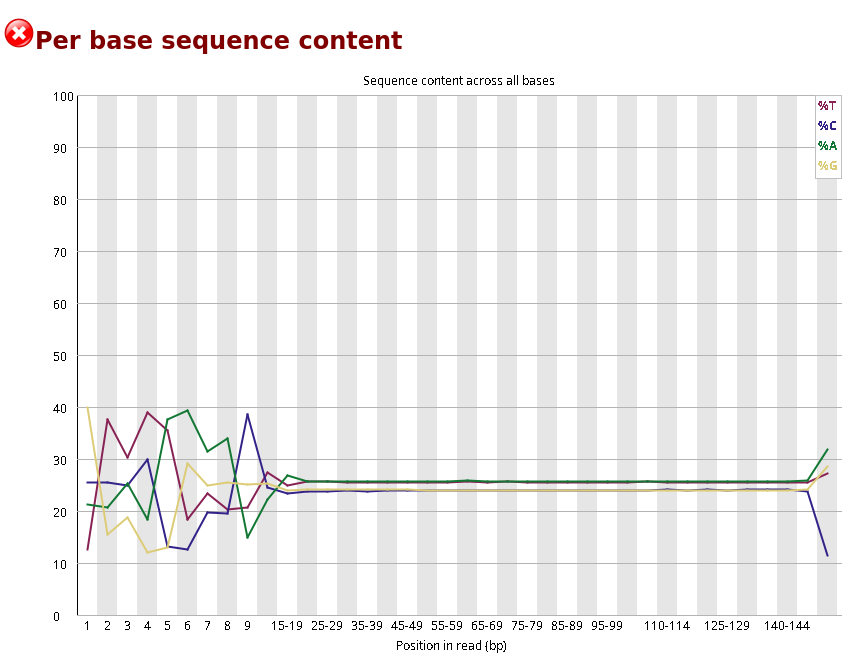
Here, we can see first 15 bases are highly fluctuating, even though they have Phred score 30. The reason is the first 10-15 bases are randomly sequenced and machines can’t capture the fluorescence properly. So, we need to trim the first few bases from each sequencing read.